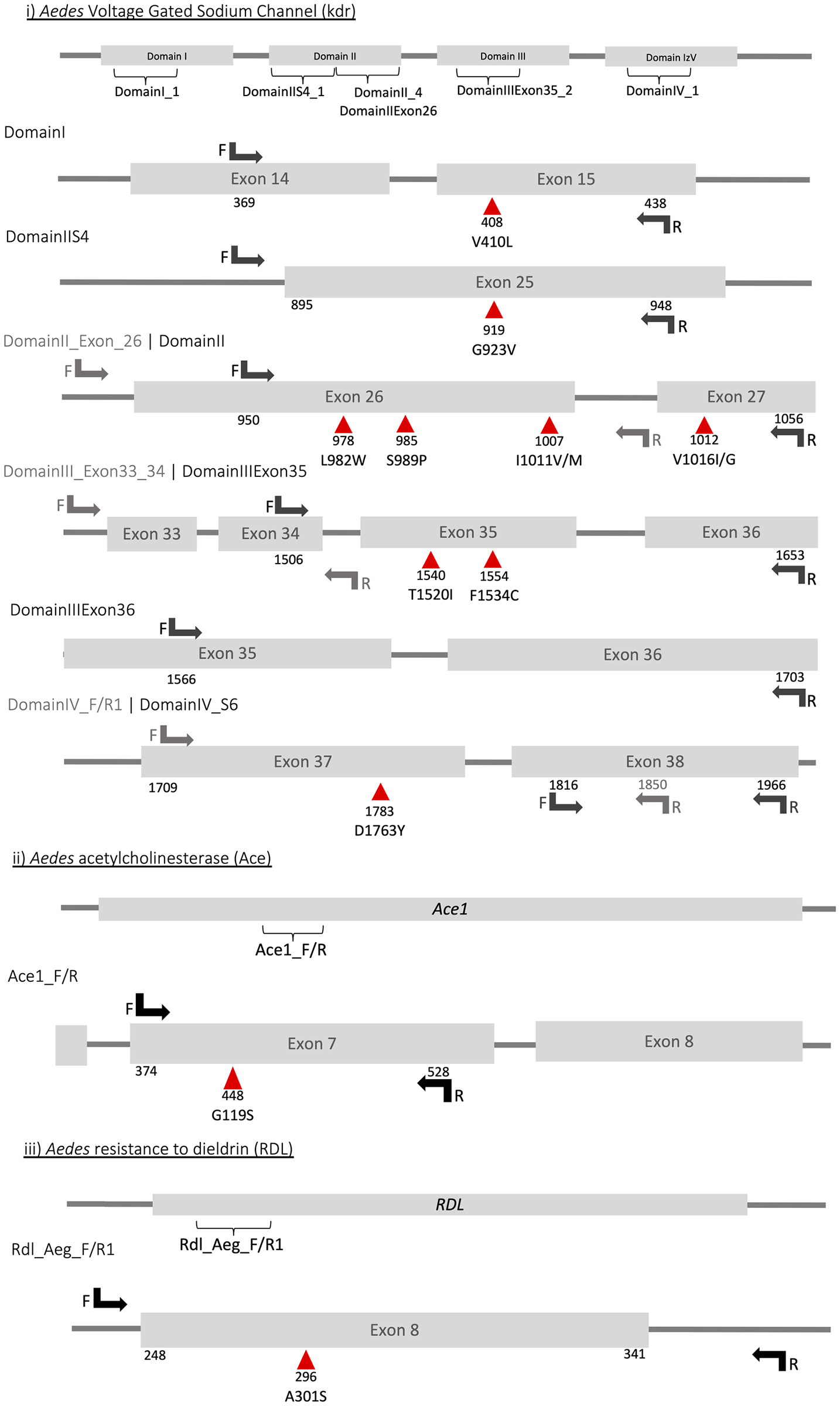
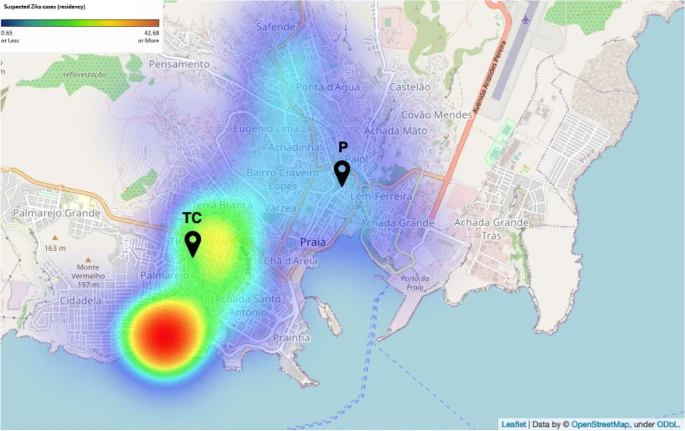
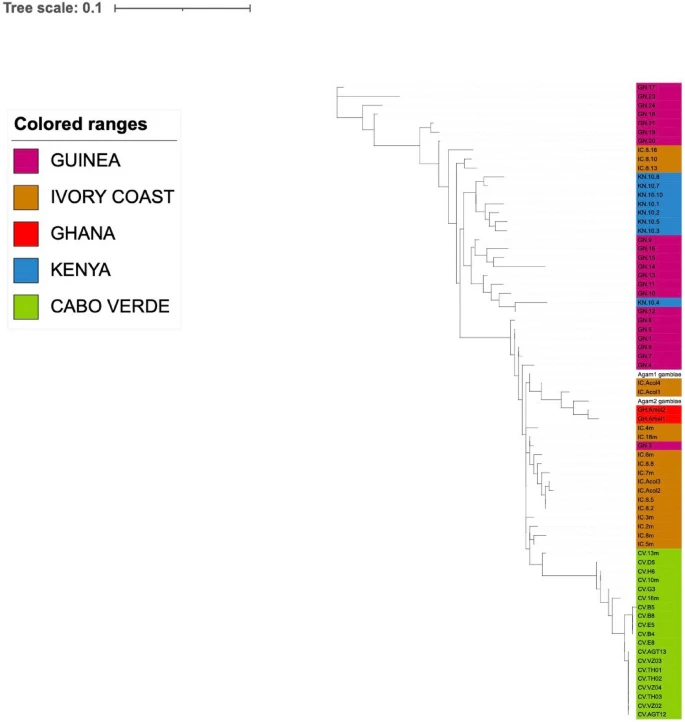
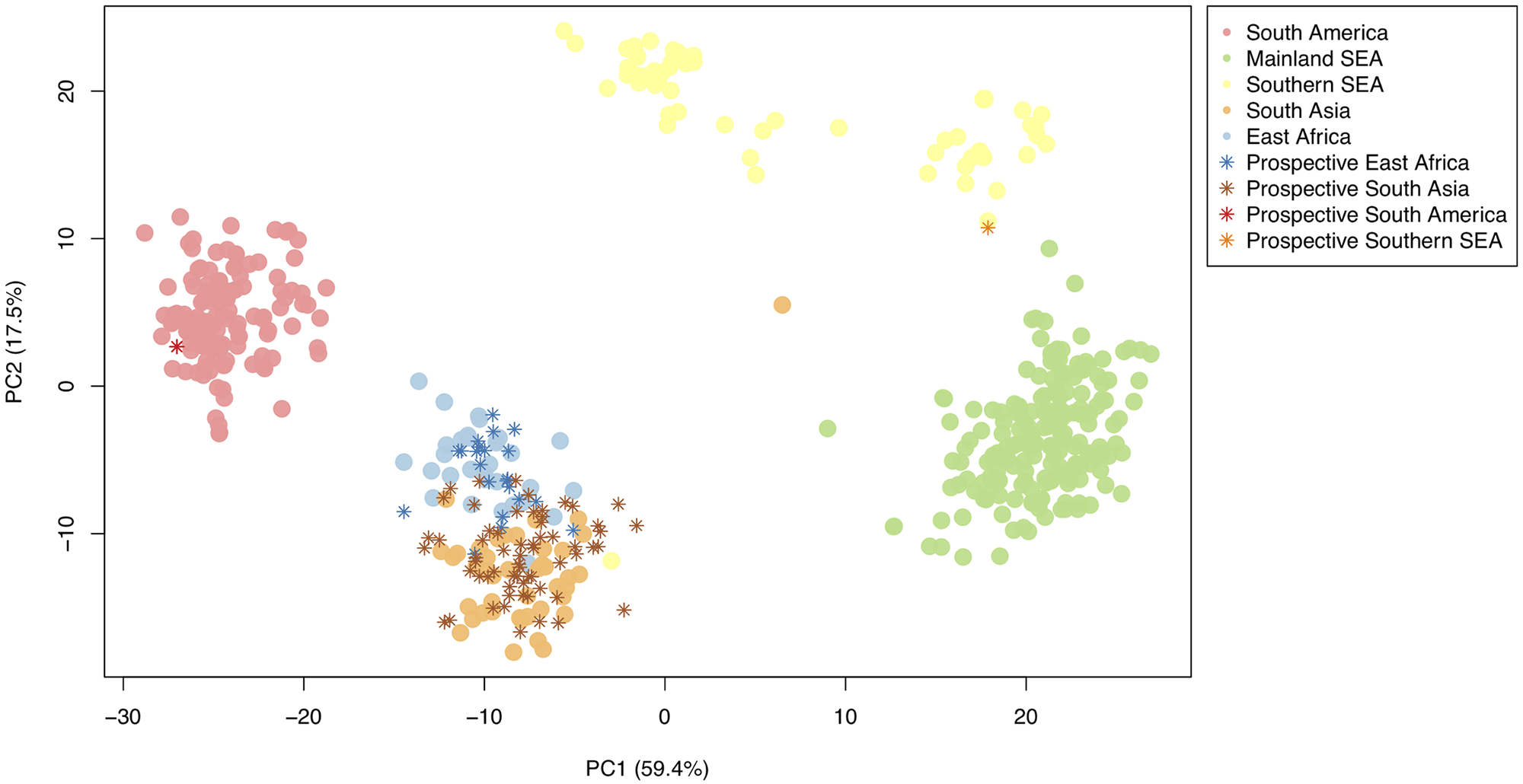
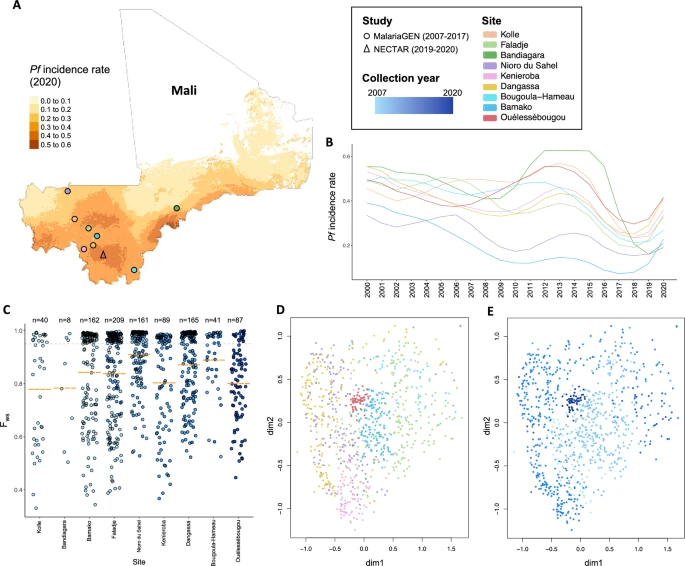
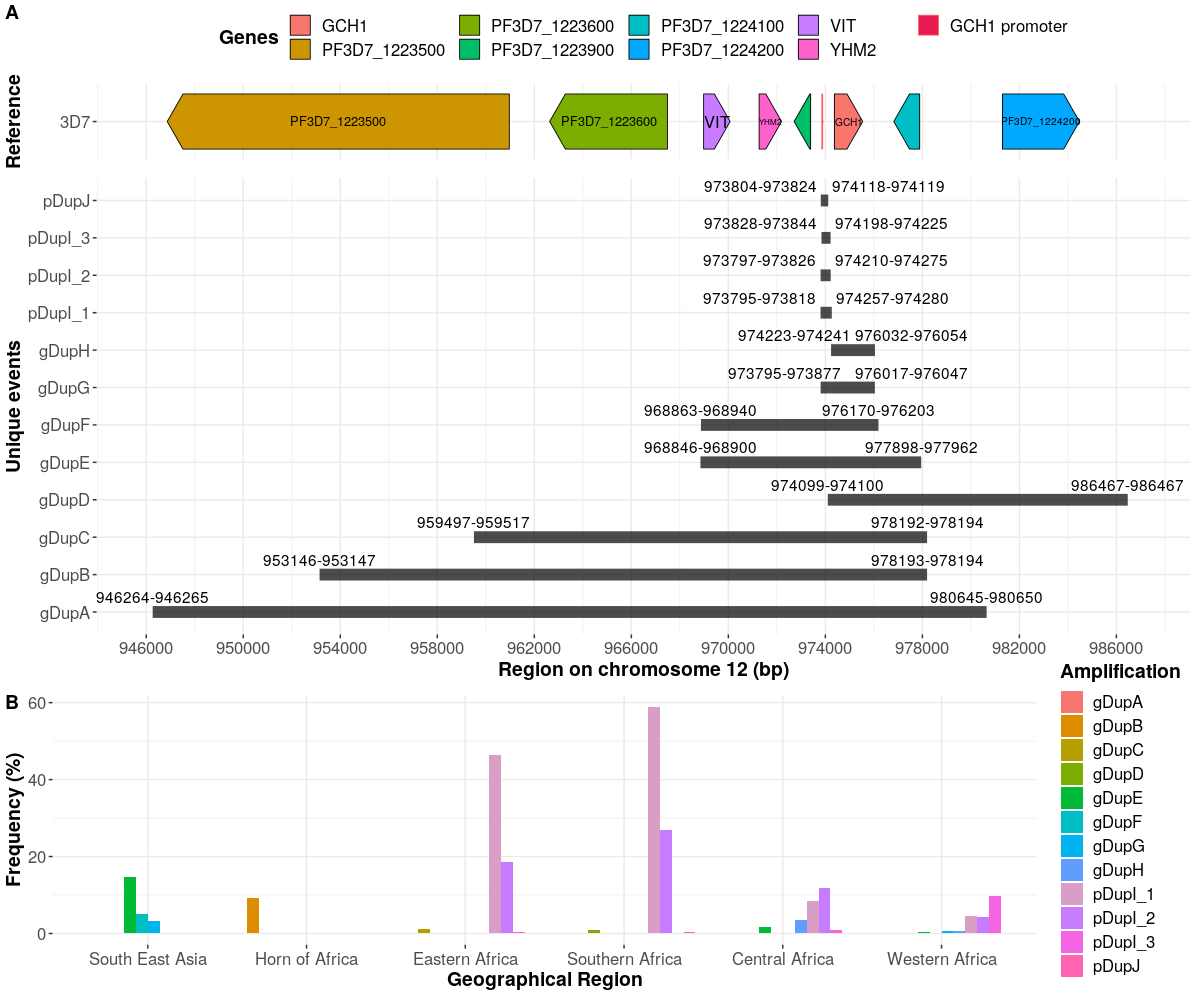
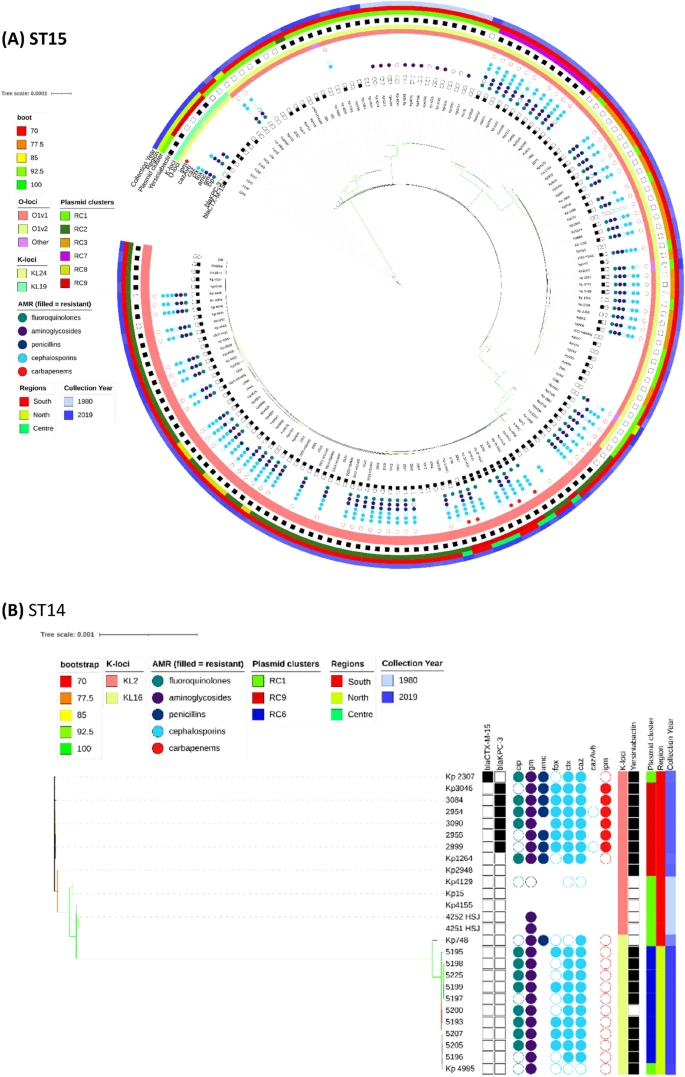
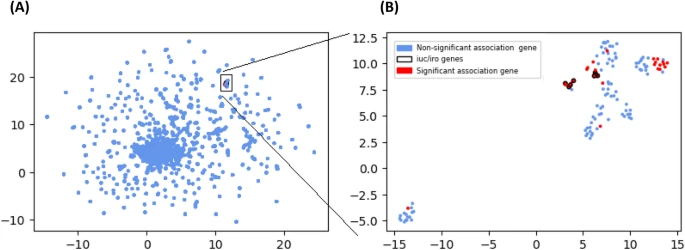
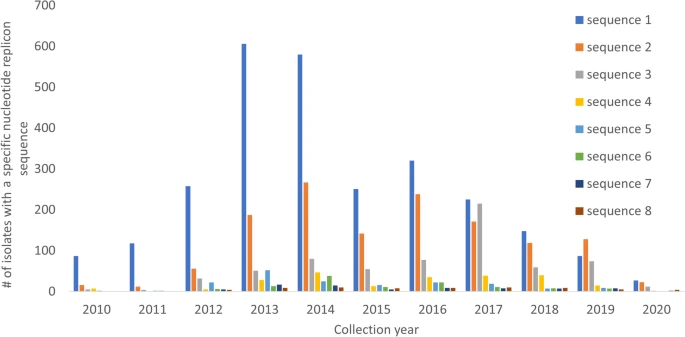
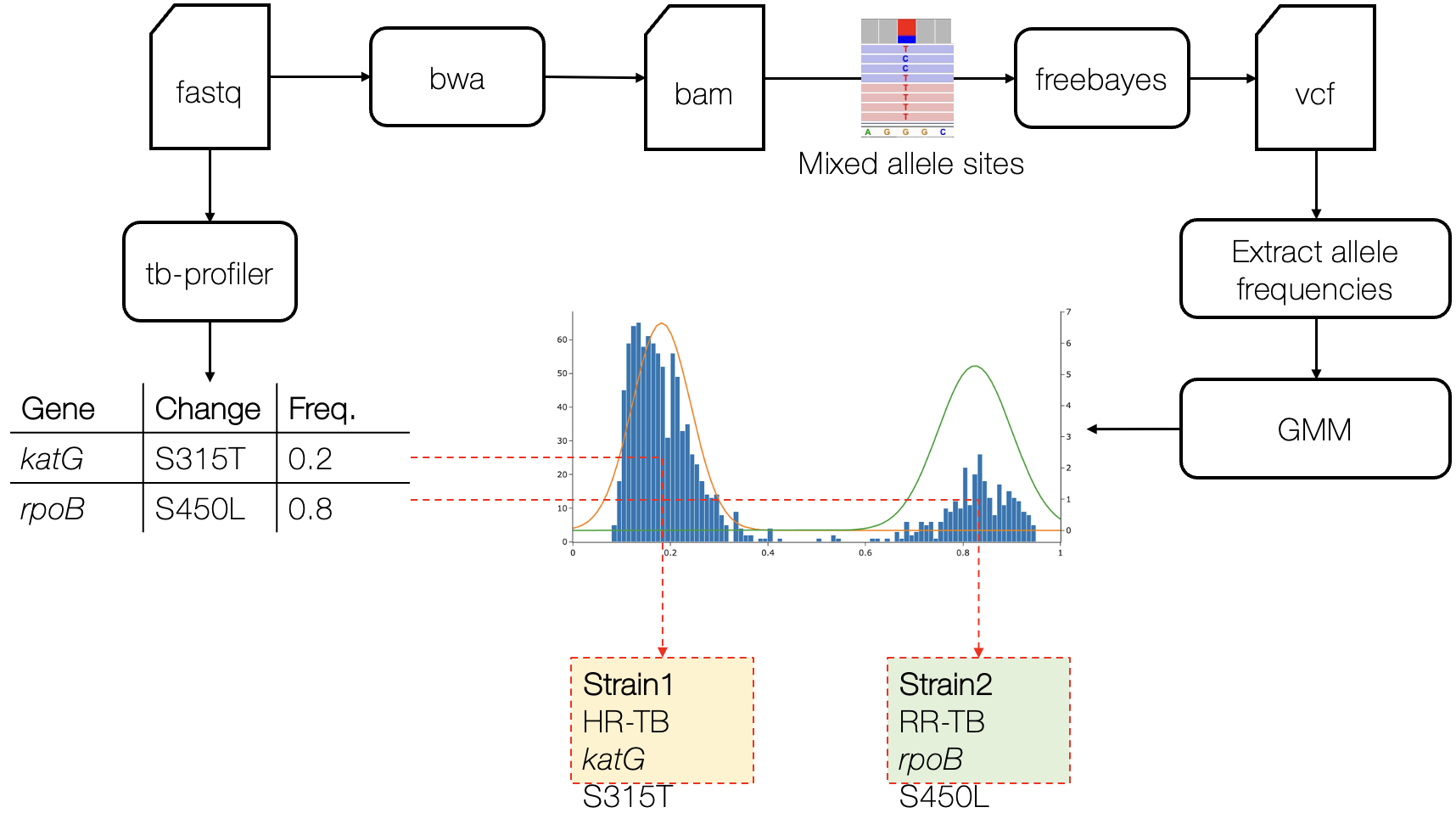
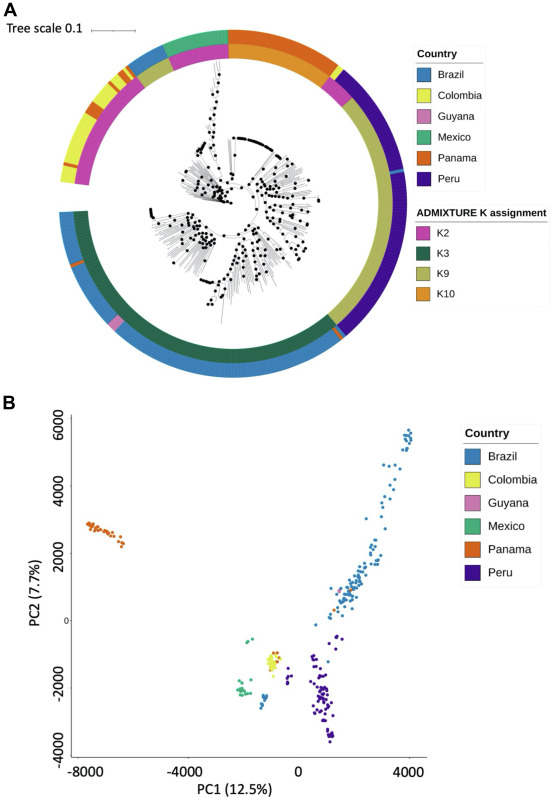
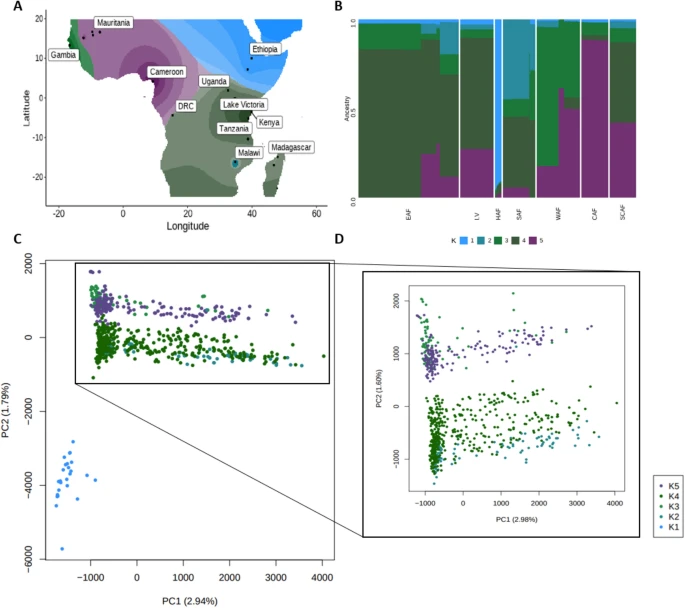
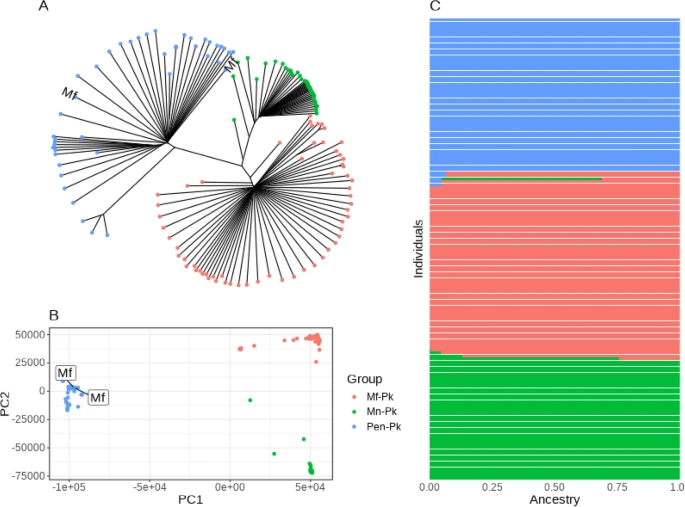
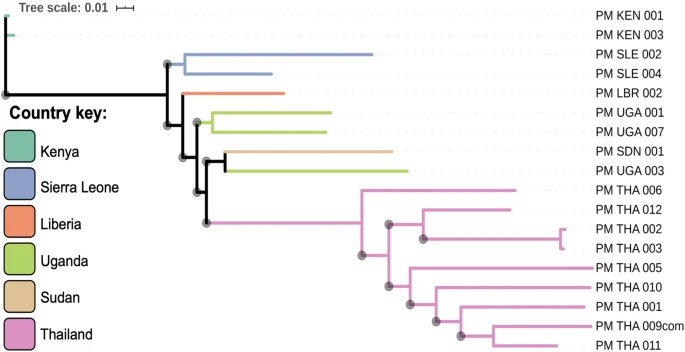
There are few population-based studies in a high prevalence area that can apply long-term large-scale whole genome sequencing. It is more challenging to interpret transmission networks when there are many possible sources of infection. Understanding transmission in these high prevalence areas would have the greatest public health benefit. The Karonga Prevention Study (KPS) in northern Malawi has been conducting research on mycobacterial infections in the region since the 1980s, with incidence of new smear positive TB around 100/100,000 and HIV prevalence is 10%. Currently, we have over 2,000 Mycobacterium tuberculosis whole genome sequences and substantial meta data available, including HIV, household membership, contact histories and GPS data, which can be used to model explicitly important questions associated with transmission, including the role of HIV and of M. tuberculosis (sub)lineage on transmissibility. We have published initial analyses from this area (Guerra-Assuncão, et al. 2015) showing decreasing transmission over time and variation between M. tuberculosis lineages 1–4, which are not confounded by host differences. By applying more advanced computational techniques to time-labelled phylogeny and transmission chain construction it will be possible to gain greater insights into molecular evolution and multiple outbreaks, including the estimation of between-patient mutation rate and ages of mutations, as well as identifying potential significant variants associated with transmission.